Agora que sabemos como quantificar a força de um ácido ou de uma base, a nossa próxima tarefa é compreender as razões fundamentais que estão por detrás de porquê Este é um grande passo: pela primeira vez, estamos a pegar nos nossos conhecimentos sobre os compostos orgânicos e a utilizar a sua capacidade de síntese. estrutura e aplicando-o a uma questão de orgânica reatividade Muitas das ideias que veremos pela primeira vez aqui continuarão a ser aplicadas ao longo do livro, à medida que abordamos muitos outros tipos de reacções orgânicas.
A: Tendências periódicas
Primeiro, vamos concentrar-nos em átomos individuais e refletir sobre as tendências associadas à posição de um elemento na tabela periódica. Utilizaremos como primeiros modelos os compostos orgânicos simples etano, metilamina e etanol, mas os conceitos aplicam-se igualmente a biomoléculas mais complexas com as mesmas funcionalidades, por exemplo, as cadeias laterais dos aminoácidos alanina (alcano), lisina (amina) eserina (álcool).
Tendência periódica horizontal da acidez e da basicidade
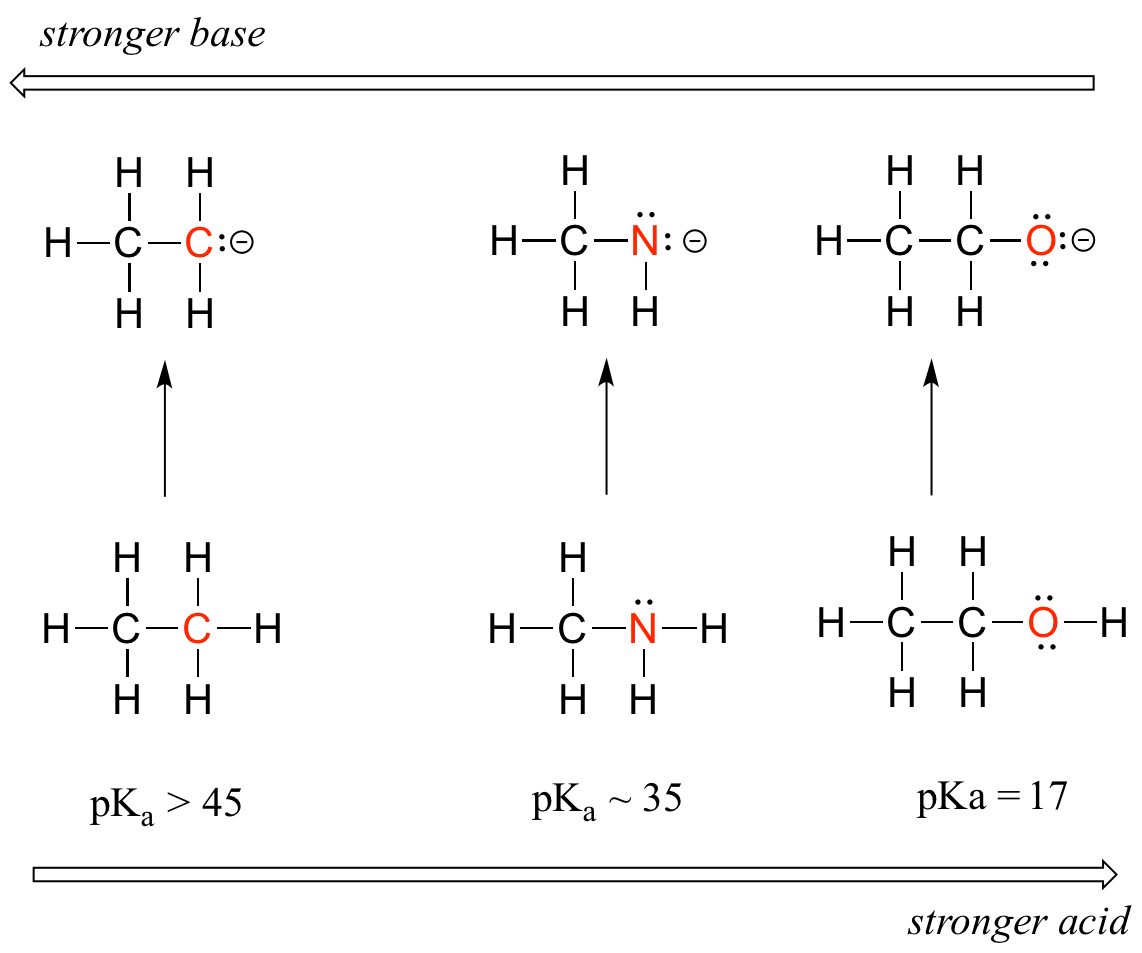
Podemos observar uma clara tendência para a acidez à medida que nos deslocamos da esquerda para a direita ao longo da segunda linha da tabela periódica, do carbono para o azoto e para o oxigénio. A chave para compreender esta tendência é considerar a base conjugada hipotética em cada caso Quanto mais estável (mais fraca) for a base conjugada, mais forte será o ácido Na base conjugada do etano, a carga negativa é suportada por um átomo de carbono, enquanto na base conjugada da metilamina e do etanol a carga negativa está localizada num azoto e num oxigénio, respetivamente. Lembre-se que a eletronegatividade também aumenta à medida que nos movemos da esquerda para a direita ao longo de uma linha da tabela periódica, o que significaque o oxigénio é o mais eletronegativo dos três átomos, e o carbono o menos.
Quanto mais eletronegativo for um átomo, maior será a sua capacidade de suportar uma carga negativa. As bases mais fracas têm cargas negativas em átomos mais electronegativos; as bases mais fortes têm cargas negativas em átomos menos electronegativos.
Assim, o anião metóxido é o mais estável (menor energia, menos básico) das três bases conjugadas e o anião carbânion etilo é o menos estável (maior energia, mais básico). Inversamente, o etanol é o ácido mais forte e o etano o ácido mais fraco.
Quando nos movemos verticalmente dentro de uma dada coluna da tabela periódica, observamos novamente uma clara tendência periódica na acidez, o que é melhor ilustrado com os haloácidos e halogenetos: a basicidade, tal como a eletronegatividade, aumenta à medida que subimos na coluna.
Tendência periódica vertical da acidez e da basicidade 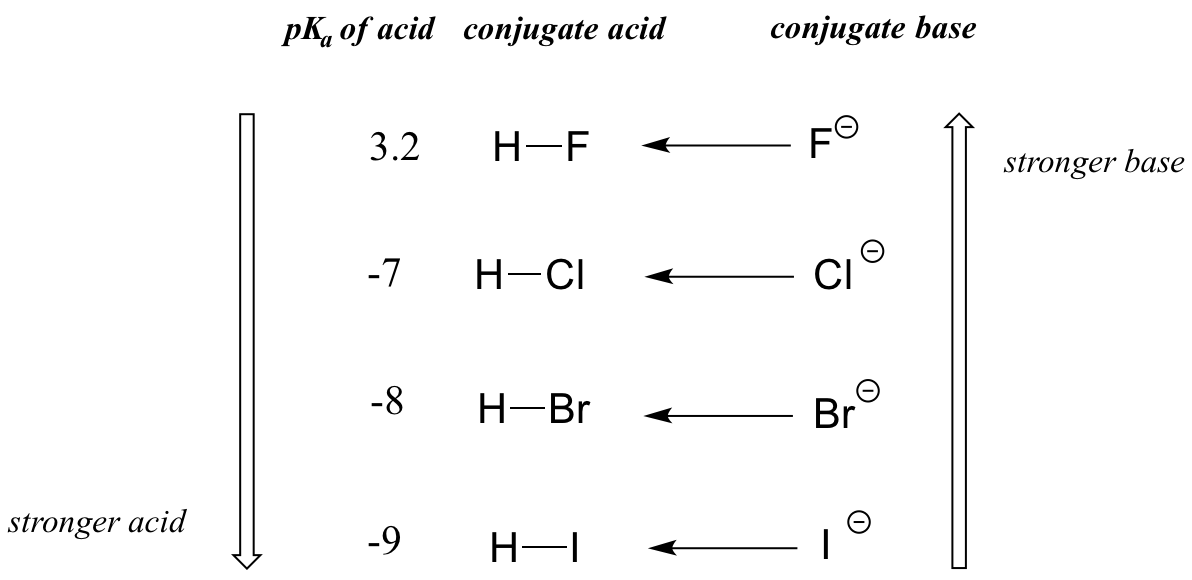
Inversamente, a acidez dos haloácidos aumenta à medida que nos deslocamos para baixo a coluna.
Para dar sentido a esta tendência, vamos considerar mais uma vez a estabilidade das bases conjugadas. Uma vez que o flúor é o elemento halogéneo mais eletronegativo, poderíamos esperar que o flúor fosse também o ião halogéneo menos básico. Mas, na realidade, é o menos Acontece que, ao mover-se verticalmente na tabela periódica, o tamanho O raio atómico do iodo é aproximadamente o dobro do raio atómico do flúor, pelo que, num ião iodeto, a carga negativa é distribuída por um volume significativamente maior:
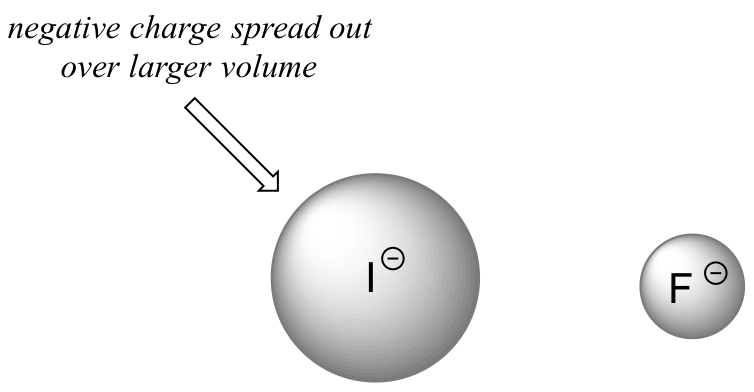
Este facto ilustra um conceito fundamental da química orgânica:
As cargas electrostáticas, quer sejam positivas ou negativas, são mais estáveis quando são "espalhadas" por uma área maior.Veremos esta ideia expressa repetidamente ao longo do nosso estudo da reatividade orgânica, em muitos contextos diferentes. Por agora, estamos a aplicar o conceito apenas à influência do raio atómico na força da base. Como o fluoreto é a menos estável (mais básica) das bases conjugadas dos halogenetos, o HF é o menos ácido dos haloácidos, sendo apenas ligeiramente mais forte do que um ácido carboxílico. O HI, com um pK a de cerca de -9, é quase tão forte como o ácido sulfúrico.
Mais importante para o estudo da química orgânica biológica, esta tendência diz-nos que os tióis são mais ácidos do que os álcoois O pK a do grupo tiol na cadeia lateral da cisteína, por exemplo, é de aproximadamente 8,3, enquanto o pK a para o grupo álcool na cadeia lateral da serina é da ordem de 17.
Lembra-se do conceito de "força motriz" que aprendemos no capítulo 6? Lembre-se de que a força motriz de uma reação se baseia normalmente em dois factores: estabilidade relativa da carga e energia total relativa da ligação. Vejamos como isto se aplica a uma reação ácido-base simples entre o ácido clorídrico e o ião fluoreto:
HCl + F- → HF + Cl-
Sabemos que o HCl (pK a -7) é um ácido mais forte do que o HF (pK a 3.2), pelo que o equilíbrio da reação se situa no lado do produto: a reação é exergónica e uma "força motriz" empurra o reagente para o produto.
O que explica esta força motriz? Considere primeiro o fator carga: como acabámos de aprender, o ião cloreto (no lado do produto) é mais estável do que o ião fluoreto (no lado do reagente). Isto explica parcialmente a força motriz que vai do reagente para o produto nesta reação: estamos a passar de um ião menos estável para um ião mais estável.
Se consultar uma tabela de energias de ligação, verá que a ligação H-F do lado do produto é mais energética (mais forte) do que a ligação H-Cl do lado do reagente: 565 kJ/mol vs 427 kJ/mol, respetivamente), o que também contribui para a força motriz: estamos a passar de uma ligação mais fraca (menos estável) para uma ligação mais forte (mais estável).
B: Efeitos de ressonância
Na secção anterior, centrámos a nossa atenção nas tendências periódicas - as diferenças de acidez e basicidade entre grupos em que o protão permutável estava ligado a diferentes elementos. Agora, é altura de pensarmos na forma como a estrutura dos diferentes grupos orgânicos contribui para a sua acidez ou basicidade relativa, mesmo quando estamos a falar de o mesmo elemento que actua como dador/acetor de protões O primeiro par de modelos que vamos considerar é o etanol e o ácido acético, mas as conclusões a que chegamos serão igualmente válidas para todos os grupos de álcool e ácido carboxílico.
Apesar de serem ambos ácidos oxigenados, o pK a O que faz com que um ácido carboxílico seja muito mais ácido do que um álcool? Tal como anteriormente, começamos por considerar a estabilidade das bases conjugadas, lembrando que uma base conjugada mais estável (mais fraca) corresponde a um ácido mais forte.
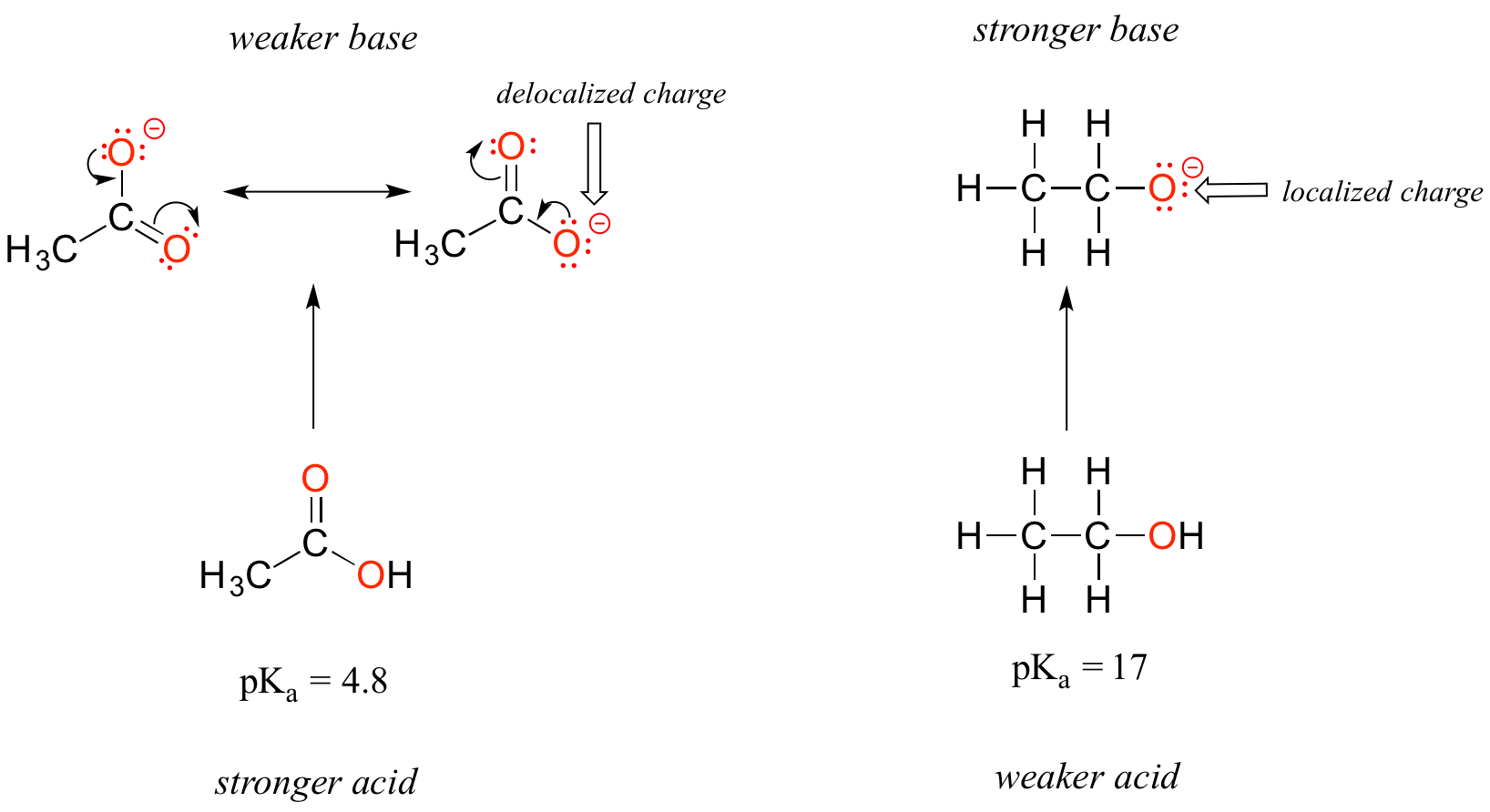
Em ambas as espécies, a carga negativa da base conjugada está localizada no oxigénio, pelo que não se podem invocar tendências periódicas. No entanto, no caso do ácido acético, existe uma diferença fundamental: podem ser desenhados dois contribuintes de ressonância para a base conjugada e a carga negativa pode ser deslocalizada (partilhada) por dois átomos de oxigénio. Em contrapartida, no ião etóxido, a carga negativa está localizada ou "bloqueada" no átomo deO ião etoxido é muito menos estável, uma vez que não tem para onde ir.
Lembre-se da importante afirmação geral que fizemos um pouco antes: "As cargas electrostáticas, sejam elas positivas ou negativas, são mais estáveis quando estão 'espalhadas' do que quando estão confinadas a um local." Agora, estamos a ver este conceito noutro contexto, em que uma carga está a ser 'espalhada' (por outras palavras, deslocalizada) por ressonância e não simplesmente pelo tamanho do átomo envolvido.
A deslocalização da carga por ressonância tem um efeito muito poderoso na reatividade das moléculas orgânicas, suficiente para explicar a diferença de mais de 12 pK a unidades entre o etanol e o ácido acético (e lembre-se, pK a é uma expressão logarítmica, pelo que estamos a falar de um fator de 1012 entre os valores de K a para as duas moléculas!)
O efeito de ressonância também explica muito bem porque é que um átomo de azoto é básico quando está numa amina, mas não Recorde-se que, numa amida, a ligação carbono-nitrogénio tem um carácter significativo de ligação dupla, devido a um contribuinte de ressonância menor, mas ainda assim importante, em que o par solitário do azoto faz parte de uma ligação pi.
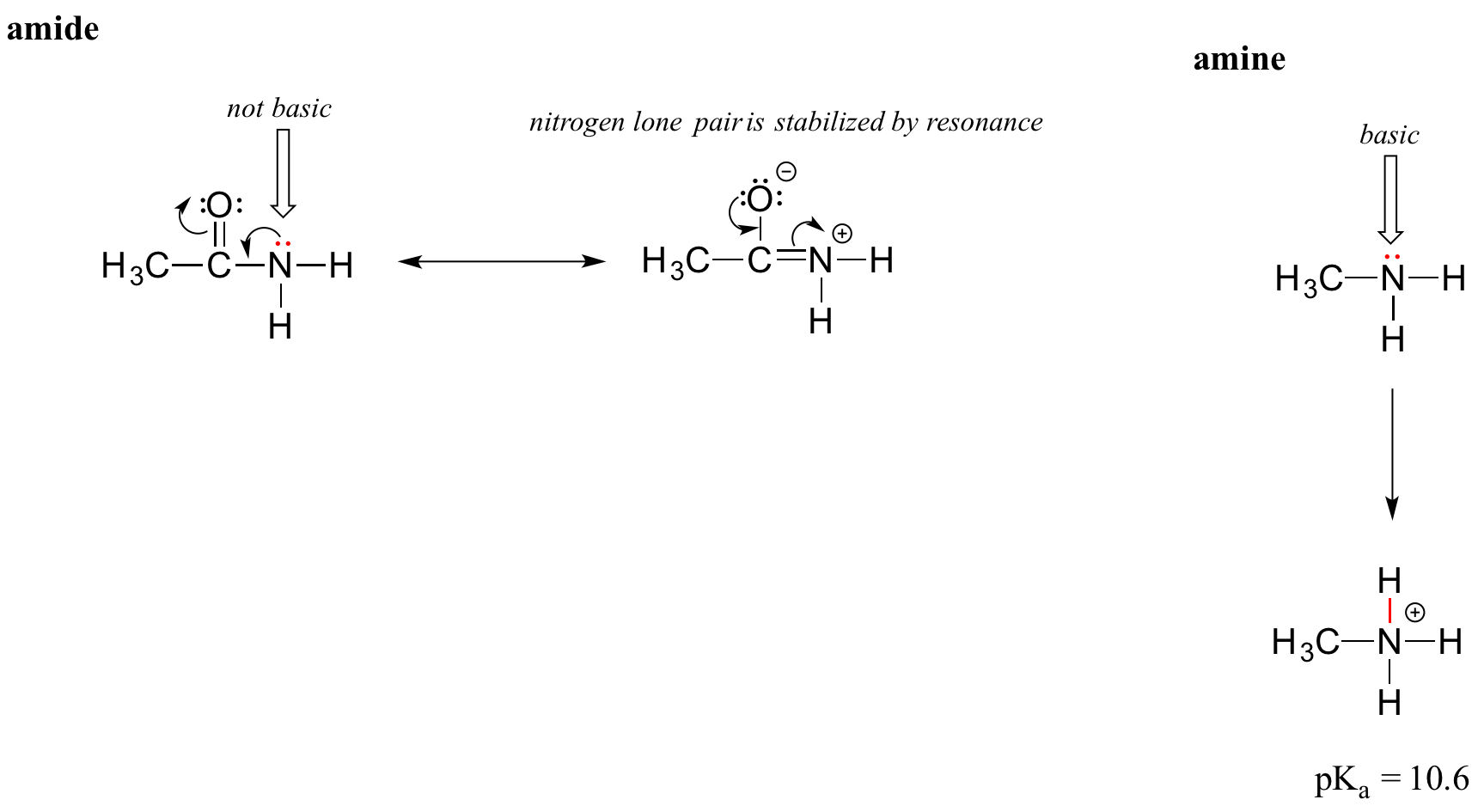
Enquanto o par solitário de um azoto amínico está "preso" num único local, o par solitário de um azoto amídico está deslocalizado por ressonância. Repare-se que, neste caso, estamos a alargar a nossa afirmação central para dizer que densidade de electrões - O par solitário de um azoto amídico não está disponível para se ligar a um protão - estes dois electrões estão demasiado "confortáveis" por fazerem parte do sistema de ligação pi deslocalizado. O par solitário de um azoto amínico, pelo contrário, não está tãoconfortável - é não parte de um sistema pi deslocalizado, e está disponível para formar uma ligação com qualquer protão ácido que possa estar próximo.
Se um grupo amida for protonado, será no oxigénio e não no azoto.